


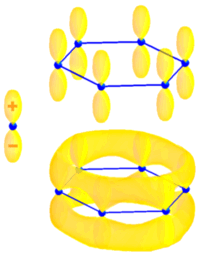
Most of the recent work on the optical microscopy and spectroscopy of single objects in based on fluorescence. Therefore it is important to recall the principal features and properties of fluorescent molecules. All fluorescent organic molecules are conjugated, i.e. they possess delocalized electronic ? wavefunctions. ? wavefunctions are built on H-atom p-orbitals, and have a node in the plane of the bond. The figure to the right illustrates this for the case of of benzene, as it shows a schematic representation of an atomic pz-orbital (left) and the lowest energy ? molecular orbital of benzene, which is constructed from a superposition of pz-orbitals centered on each of the six carbon atoms. The ?-electron can be seen as “moving” in a torus following the conjugation path of the molecule. This very simple picture is known as the 1D electron gas model.
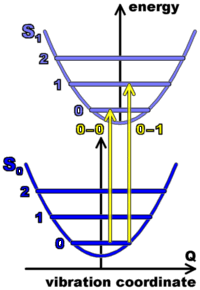
When the electronic cloud of a molecule is excited (after the absorption of one photon), the ?-electron density is changed, which means that the geometry of the molecule, in particular the distances between the carbon atoms, is slightly changed. This leads to coupling of the electronic excitation with vibrations. Therefore, absorption leads to an electronic transition accompanied by a series of vibronic bands where 0, 1, 2, etc. vibration quanta are created along with the electronic excitation. The figure on the left side shows potential energy curves for this vibration of the atoms in the ground and first excited state of the electron cloud, as functions of a vibration coordinate. Absorption induces transitions between levels, as indicated by the arrows.
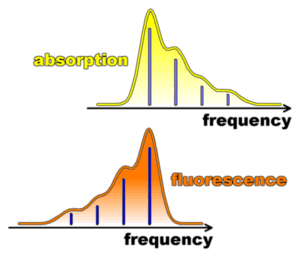
In the conjugated fluorescence process, one photon is emitted from the relaxed excited state (0 vibrations) to vibrational levels of the ground state. Vibronic coupling will give rise to side-bands at higher energy than the 0-0 transition in the case of absorption, to side-bands at lower energy in the case of fluorescence. For purely harmonic vibrations, this leads to symmetry (mirror image) between normalized absorption and fluorescence spectra as a function of energy (or frequency). A schematic representation of the absorption and the fluorescence spectrum of a molecule as a function of the frequency is shown on the right. The strengths of the respective transitions are represented by sticks, or by very sharp lines at low temperatures. At room temperatures, all lines are broadened (smooth contours).
It is important to consider how strongly a molecular transition is coupled to radiation. The quantity describing the strength of this coupling is called oscillator strength. It is proportional to the square of the transition dipole moment, a vector homogeneous to an electric dipole and fixed relative to molecular axes. Being conjugated processes, absorption and fluorescence strengths are related (inasmuch as the geometry of the molecule does not change too much between absorption and emission). The inverse of the radiative lifetime of the molecule is proportional to the integral of its absorption coefficient over the spectrum (Strickler-Berg relation). However, other channels than radiative can be accessible from the excited state of the molecule, thus decreasing the fluorescence intensity. The ratio of the radiative rate and the sum of the radiative and the nonradiative rate is called the fluorescence quantum yield. Molecules with high radiative rates will therefore tend to be better emitters. This is the case of many laser dyes, who are also good fluorescent probes.
The non-radiative channel is mainly governed by radiation-less transitions to lower-lying electronic states, in particular the ground state, and to a lesser extent the metastable triplet state (see below). Non-radiative transitions are much reduced for planar, rigid molecules. As a rule, therefore, good fluorophores have to be planar, rigid and sufficiently stable chemically to sustain many excitation-emission cycles in such reactive environments as air or water. The latter is particularly important for biological labelling.
A schematic level scheme of an organic molecule which includes the three main electronic states involved in absorption, fluorescence, and intersystem crossing is shown below. (Such a scheme is called a Jablonski diagram)
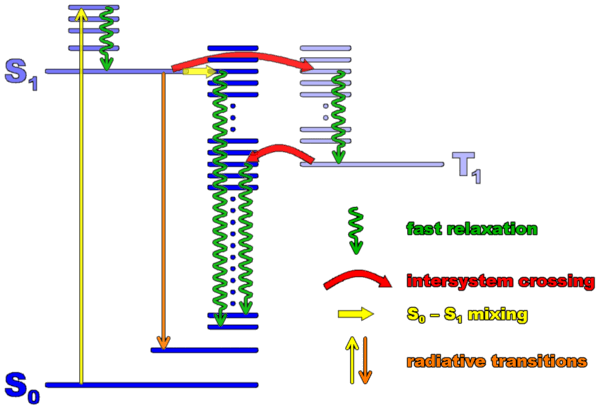
The ground state of a large majority of fluorescent molecules is a singlet state, i.e. their electrons are paired into a zero spin states. The ground state is called S0. The first excited singlet state S1 is the one reached after absorption of a photon. The reason for this is that a photon cannot flip spins (because magnetic field effects are weak). But there is another excited state at lower energy (because of exchange interaction) than the first excited singlet. It is the triplet state T1, with total spin of 1. It therefore has three spin sublevels (usually undistinguishable at room temperature). Transitions between singlet and triplet states are called intersystem crossing (ISC). ISC is spin-forbidden, therefore caused only by weak interactions such as spin-orbit coupling. ISC transition rates are rather low accordingly, and the lifetime of the triplet state is rather long (often microseconds to seconds). However, the triplet states play a central role in the photodynamics of the molecule, because they limit the number of resonant absorptions and fluorescence per unit time that a molecule can perform.
It must be realized that aromatic molecules, being conjugated, are rather reactive : the ?-electrons are very polarizable (being far from the carbon nuclei), and therefore readily engage in new chemical bonds. The situation is even worse when the molecule is excited, because then the electronic cloud has been given extra energy, which can be used to cross photochemical barriers, or gives access to more new states for the excited electron. Fluorescent molecules, which have long excited state lifetime, are therefore particularly sensitive to photochemistry. Among the possible reactions taking place in excited molecules, we can cite: charge (electron or hole) transfer, proton transfer (intra- or intermolecular), cis-trans isomerization of double bonds, twisted intramolecular charge transfer, photooxidations, photoadditions, etc. The immense majority of these reactions destroy fluorescence. The new product does not absorb light in the same spectral region, or if it absorbs, does not emit any more, because of new non-radiative channels. All fluorescent molecules are therefore subject to photobleaching, that is the “death” of fluorescence after some excitation-emission cycles. This number can vary between less than 1 and several hundreds of millions, depending on the molecules themselves, atmosphere, temperature, solvent, and a number of other conditions.
Fluorescence is a very low background technique. The reason is that fluorescence arises only after real excitation of the molecule in its excited state. If a molecule is not excited resonantly, it may still end up in an excited vibrational level of the ground state: this is a Raman scattering process. As compared to fluorescence, the probability of this process is reduced by the extremely short dwell time in the “virtual” excited state. Raman scattering cross-sections are of the order of 10-12 Å2, i.e. more than 10 orders of magnitude smaller than the typical fluorescence cross-section of a good fluorescent dye, 3 Å2. This explains why spectral selection by fluorescence is so efficient.